
.png)
Machine learning models are helping researchers predict how a potential drug molecule might "dock" with a target protein in the body. But a recent paper in the Journal of Cheminformatics from researchers at Recursion finds that these models often focus on how a molecule is positioned while overlooking its ability to make precise protein-ligand interactions. Missing this critical information leads to a gap between academic benchmarks and real-world drug design.
While the paper highlights the need for improved protein-ligand interaction modeling in AI models, the field is rapidly evolving in that direction, says David Errington, Senior AI Research Scientist at Recursion, and one of the study’s authors. New models like Boltz-2 – developed by Massachusetts Institute of Technology in partnership with Recursion – are already making enormous progress in modeling not only protein structure, but protein binding affinity, and catching up to traditional physics-based modeling.
“This is new, exciting technology,” says Errington, “and we’ve already seen huge progress in the past 18 months – but there’s still work to do.”
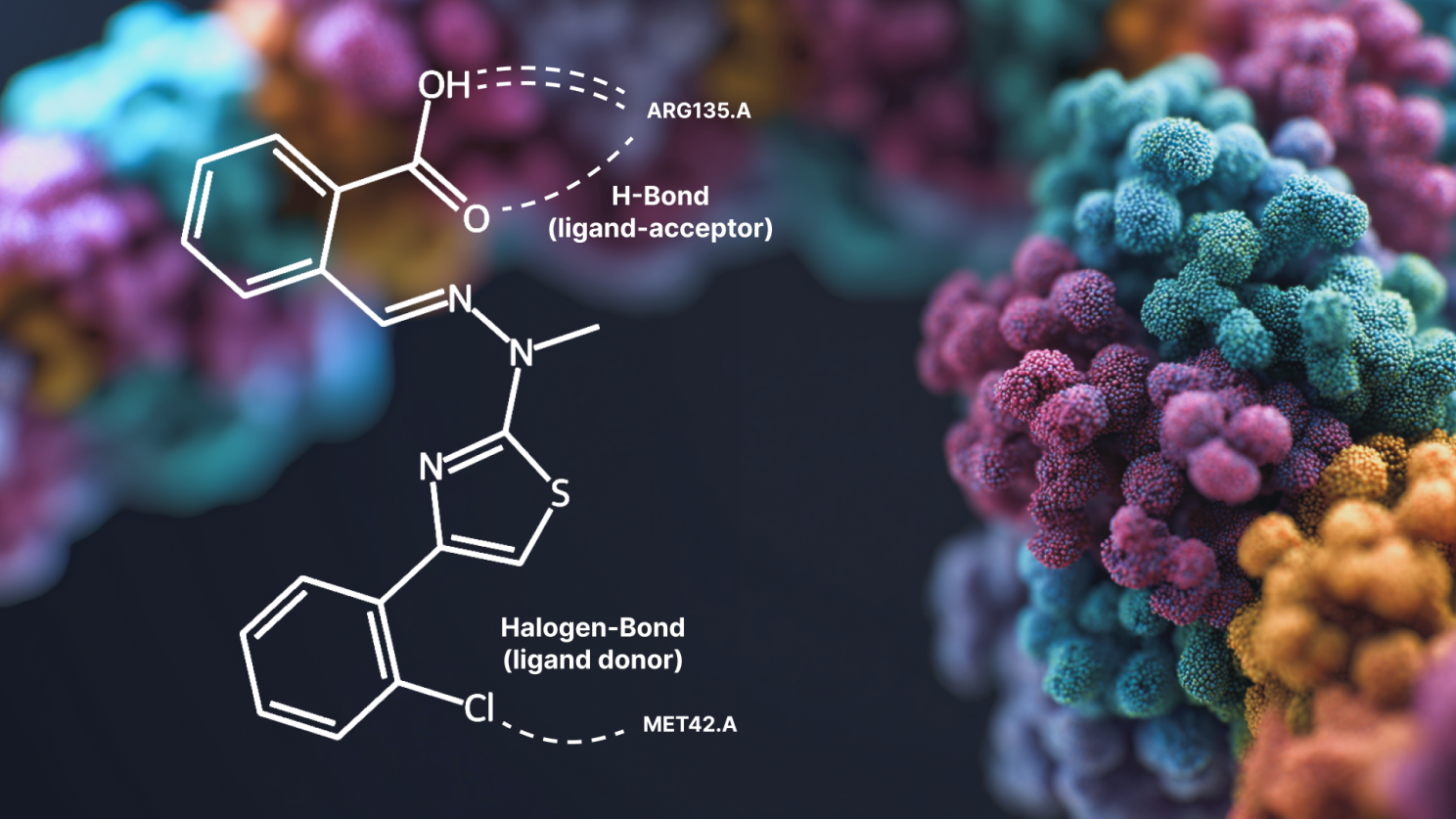
The study highlights that while newer ML cofolding models perform well at predicting the 3D position (or "pose") of a drug molecule in a protein pocket, they often fail to replicate key chemical interactions, like hydrogen bonds, that are an essential part of structure-based drug design.
The researchers used PoseBusters to evaluate several pose-prediction tools – including both classical methods and newer ML methods. By considering the recovery of protein-ligand interactions, they demonstrate that conventional docking metrics can often overestimate model performance, particularly in ML methods.
Specifically, they found that:
Although ML methods still lag behind classical methods in terms of their understanding of protein-ligand interactions, they have the capacity to offer significant improvements to traditional approaches as their performance continues to improve.
Unlike traditional methods, ML models can enable structure-based drug design in the absence of a crystal structure. Creating a crystal structure of one protein is enormously time- and cost-intensive, says Errington – often “an entire PhD’s worth of work.” Furthermore, he adds, with ML models, the protein is able to fully adapt to accommodate very different ligands – something that would be difficult to achieve in a classical docking campaign.
“This is definitely the direction of travel in the field,” he says, “we just need to ensure we encode the physics correctly."
Boltz-2 was designed to tackle the “binding affinity problem” that plagues early stage AI drug discovery – providing a means to quickly and efficiently estimate the absolute binding free energies (ABFE) of small molecules to proteins without relying on experimental crystal structures. Until now, determining binding affinity computationally has been time- and cost-intensive – requiring atomistic, physics-based simulations like free-energy perturbations, which has represented a major bottleneck in early stage drug discovery.
“Boltz-2 shows real progress when examined under this lens of protein-ligand interaction recovery,” says Errington.
-------------------------
Author: Brita Belli, Senior Communications Manager at Recursion.